We are happy to announce the release 4.0 of gene.iobio, our real-time variant prioritization web app. This is a complete overhaul with richer variant annotations, a streamlined user interface, and a powerful new backend.
Let’s walk through the biggest improvements.
A focused, easy-to-understand user interface.
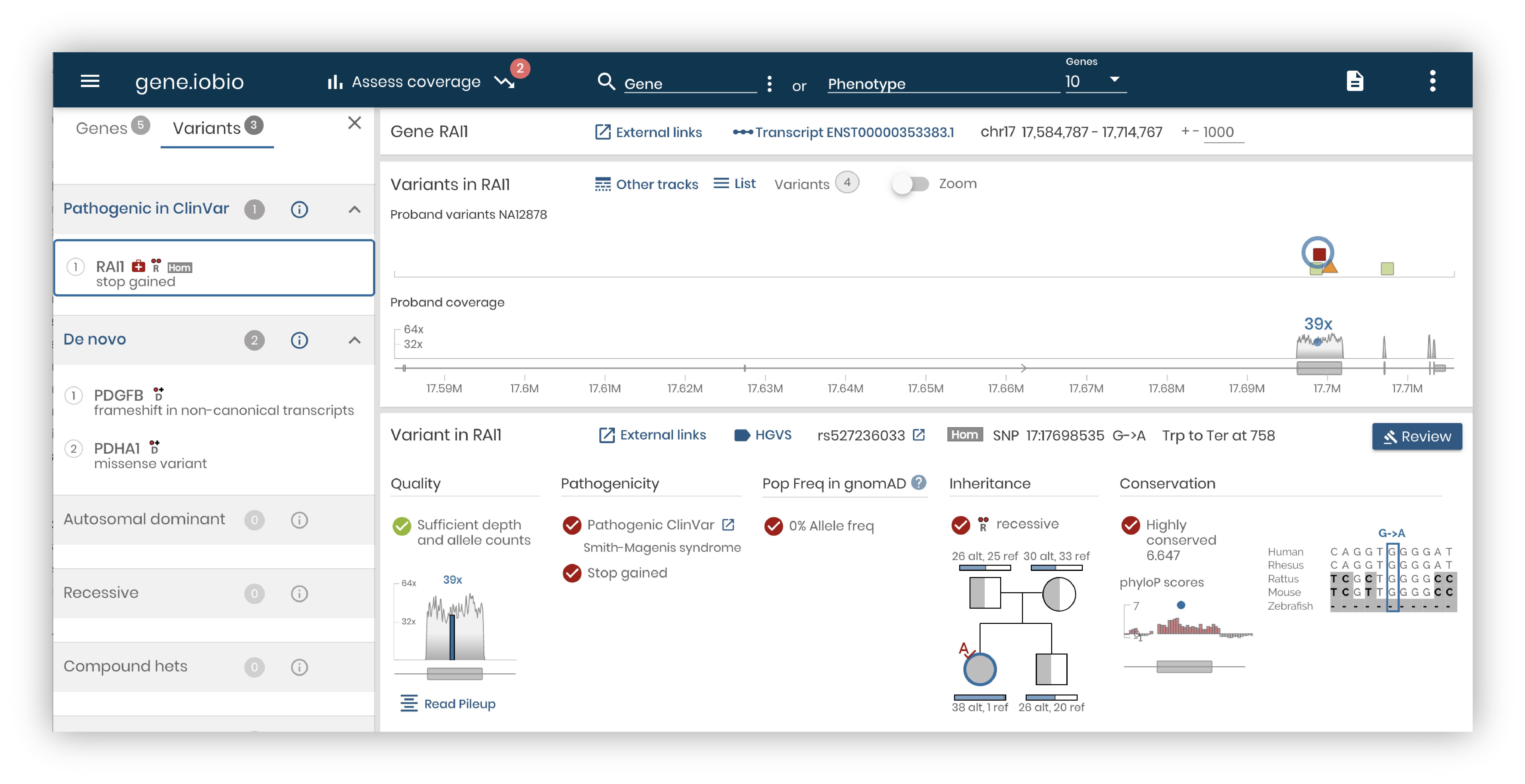
Our user interface design seeks to answer this question. How do we convey the most important information for variant discovery without sacrificing context and missing out rich annotations from different public resources? In past releases of the app, the navigation and the main panels centered around genes. But as we watched how researchers and clinicians used our app, we realized that variants needed to drive the analysis.
To achieve this, we redesigned the Variant Detail panel, organizing the data and the visualizations around answering these critical questions:
- Is the variant real?
- Is the variant pathogenic?
- Is the variant sufficiently rare?
- Does the variant follow the expected Mendelian inheritance pattern for this family?
- Is this variant within a conserved genomic region?
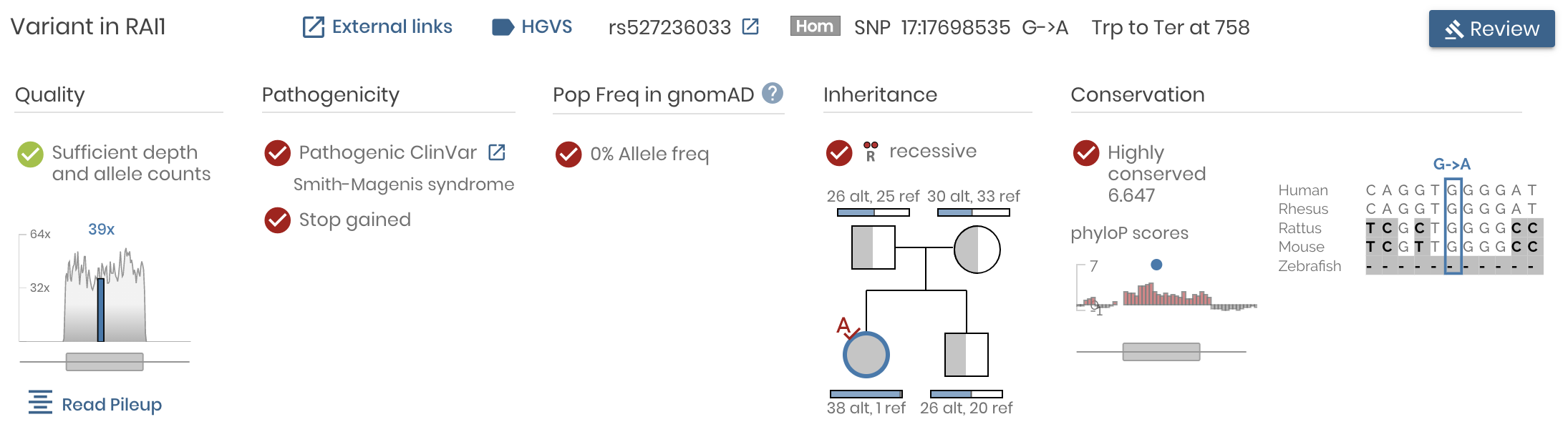
At the same time, we made it easy to explore the underlying data from different angles. Click on the “Read Pileup” button to examine IGV read level alignments of the variant and surrounding region. Click on “External Links” to launch the UCSC browser, GTex, VarSome, gnomAD, ClinVar and many more helpful external resources.
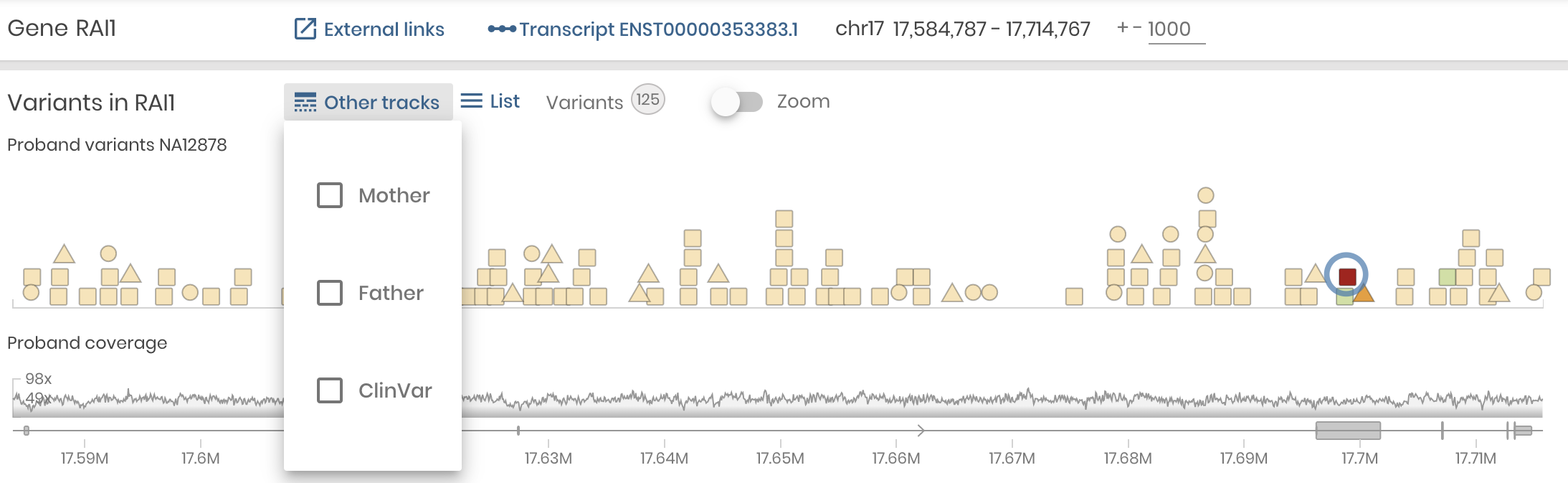
To make Variant Detail the main focus, we made another important shift. We simplified the Gene panel and we made the Variants Panel a supporting player. Now, it will show the color coded variant in the gene along with gene coverage, just as it did before, but only for the Proband. Other variant tracks can be displayed on demand, including the Mother and Father in a Trio study and the ClinVar track showing variants of user selected pathogenicity catalogued for this gene.
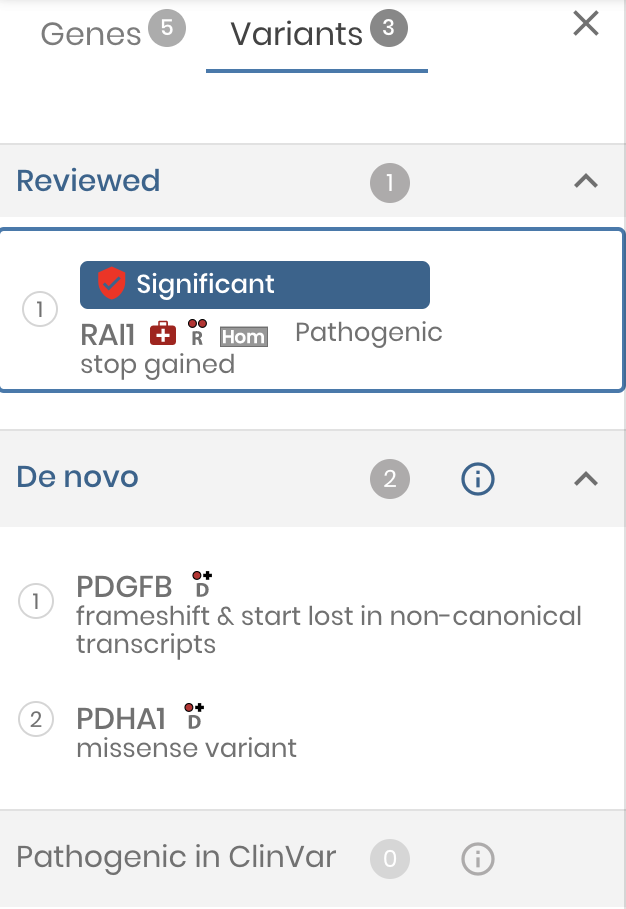
Variant review
We have taken big steps to make gene.iobio a genomic workbench for variant review. Now variants can be assigned an ‘interpretation’ and notes can be attached.

As variants are reviewed, the left variant panel serves as the main navigation, conveniently showing the reviewed variants at the top of the list.
Gene.iobio is fully integrated with Frameshift’s Mosaic https://frameshift.iobio/, a collaborative platform to organize and explore genomic data. Pairing gene.iobio with Mosaic makes start-to-finish variant prioritization a reality.
A new backend
The research community requires genomic data be served from institutional servers or virtual private cloud instances. Although local deployments were possible with our micro-service architecture, it could be a challenge to install and maintain in different compute environments. So we re-engineered our backend with the goal of minimizing system dependencies while still supporting a scalable, secure service architecture. Thanks to iobio team member Anders Pitman for this huge overhaul!
More to come
In future releases, we have plans to take full advantage of ClinVar annotations, mining and visualizing the clinical phenotypes of the catalogued pathogenic variants for a gene. We will continue to make performance improvements so that gene.iobio integrates, annotates, and visualizes information in real-time.


