Gene.iobio accesses variant and sequence alignment files to perform real-time analysis. This blog post explains how to load your data files.
File formats
VCF file
The main input to gene.iobio is the variant file. You will need access to a VCF file that has been compressed and indexed. If you have a VCF file, but it has not been compressed and indexed, you can learn more in the blog post Compressing and indexing VCF files. The app will need access to both the compressed VCF and the index file. For example, the demo variant data shown in gene.iobio uses these files:
platinum-exome.vcf.gz platinum-exome.vcf.gz.tbi
BAM file (optional)
The other input to gene.iobio is the sequence alignment file, using the BAM format. When provided, the sequence alignment files are used in the app to analyze coverage and call variants on-demand for genes of interest. These are very large files and normally are stored in this binary form. The app will need access to both the BAM file and its index file. For example, the demo sequence alignment data for the proband uses these files:
NA12878.exome.bam NA12878.exome.bam.bai
Occasionally, you might have access to the BAM files, but not the VCF files because the pipeline has yet to complete the variant calling step. No problem. You can load the BAM file(s) without the VCF files and the app will automatically call variants.
Bookmarked Variants file (optional)
Output from gene.iobio is stored in a comma separated or tab separated file. This file contains any variants that have been bookmarked in the app and represent the variants of interest that are being evaluated. Please see the gene 2.3.0 blog post to learn more about bookmarking variants. There is also a Saving your Analysis video that walks you through this functionality.
Where is your data stored?
You can load data files into gene.iobio by either accessing the files from your local drive or from a URL if the files are accessible from a web server. For example, the demo data is stored on an Amazon S3 bucket, so the URL for the variant file looks like this:
https://s3.amazonaws.com/iobio/samples/vcf/platinum-exome.vcf.gz
In this case, the index file is stored in the same bucket.
https://s3.amazonaws.com/iobio/samples/vcf/platinum-exome.vcf.gz.tbi
But if the index file is available from a different URL path, you can check the button ‘Separate URL for index files’ to specify the URL to the index file.
No lengthy upload required
Gene.iobio streams data to the backend services in gene-sized chunks, so there you can start analyzing your data as soon as the files have been specified.The Files dialog
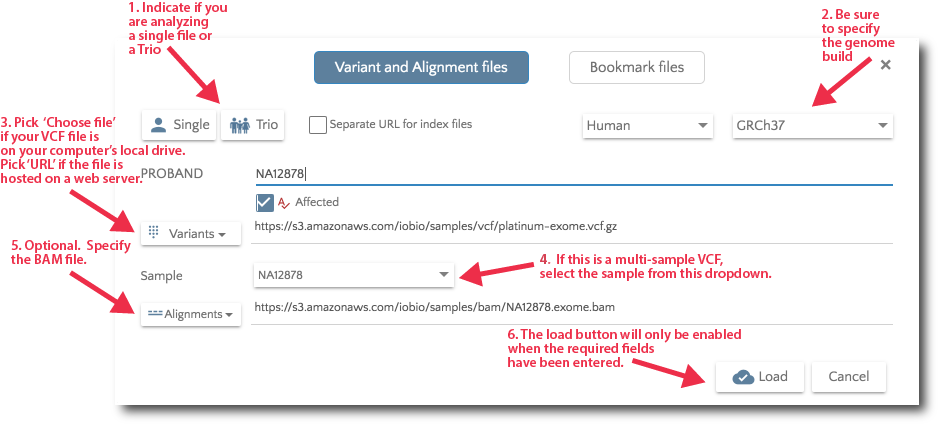
To specify your data files, click on the ‘Files’ link in the nav bar.

Specify your data files from the Files dialog.
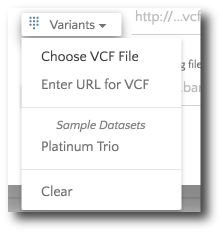
If your files are stored locally, selected ‘Choose VCF file’ from the ‘Variants’ dropdown.
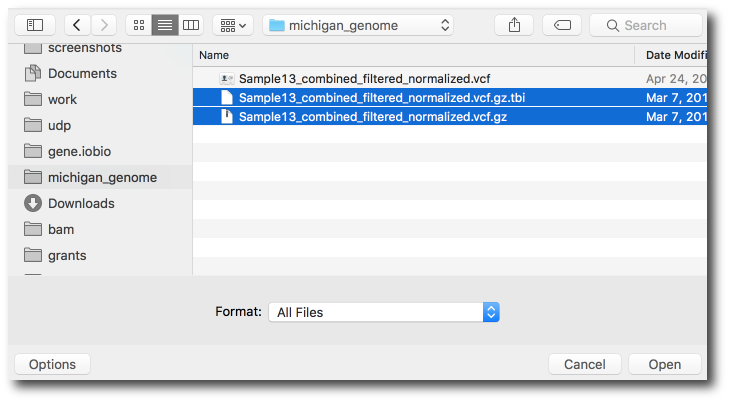
Then multi-select both the vcf.gz and the vcf.gz.tbi files.
For Trio analysis, specify the files for Mother and Father. Additionally, you can specify affected and unaffected sibs.
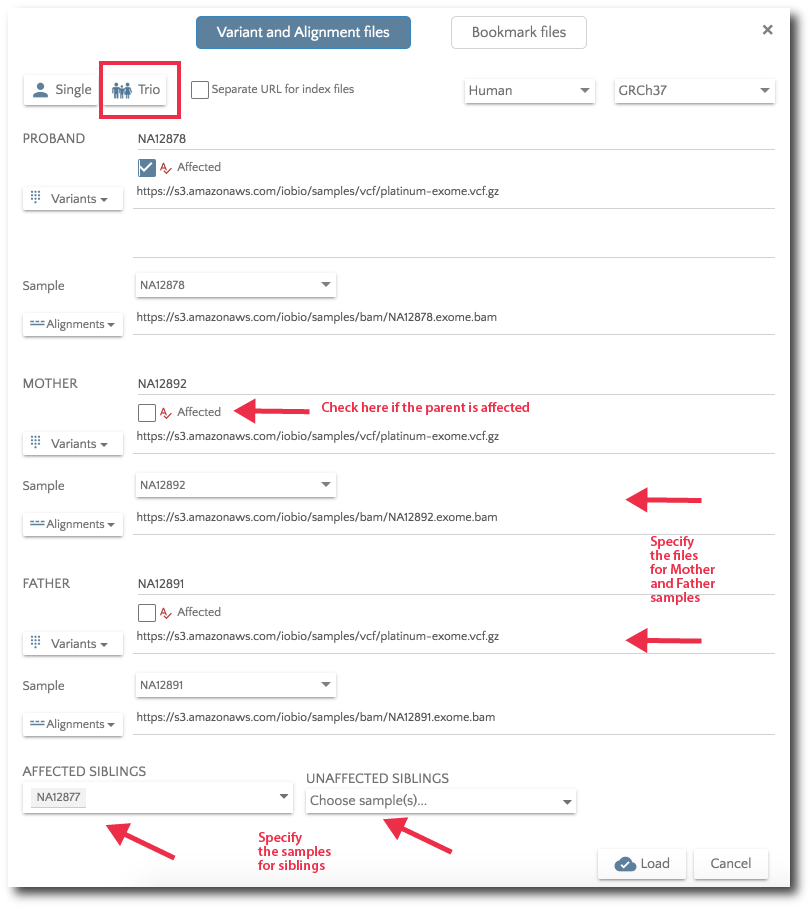
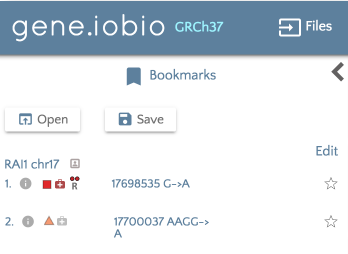
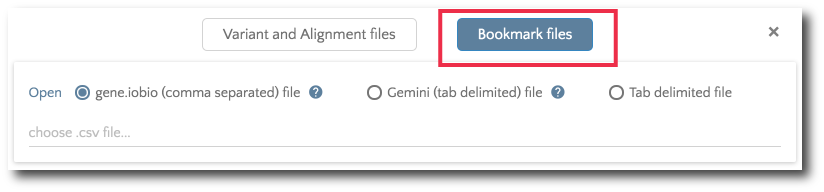
If you would like to load previously bookmarked variants, click on the ‘Bookmark files’ button and then specify the file format.
After the file is selected, the bookmarks will load and show in the left hand panel.