The first app in the iobio family, bam.iobio, has had a facelift, and is now better than ever. As a demonstration, I am going to take a look at my own genome to highlight a couple of the changes. This demonstration will also show how well iobio apps play together, acting in concert to help answer interesting genomic questions.
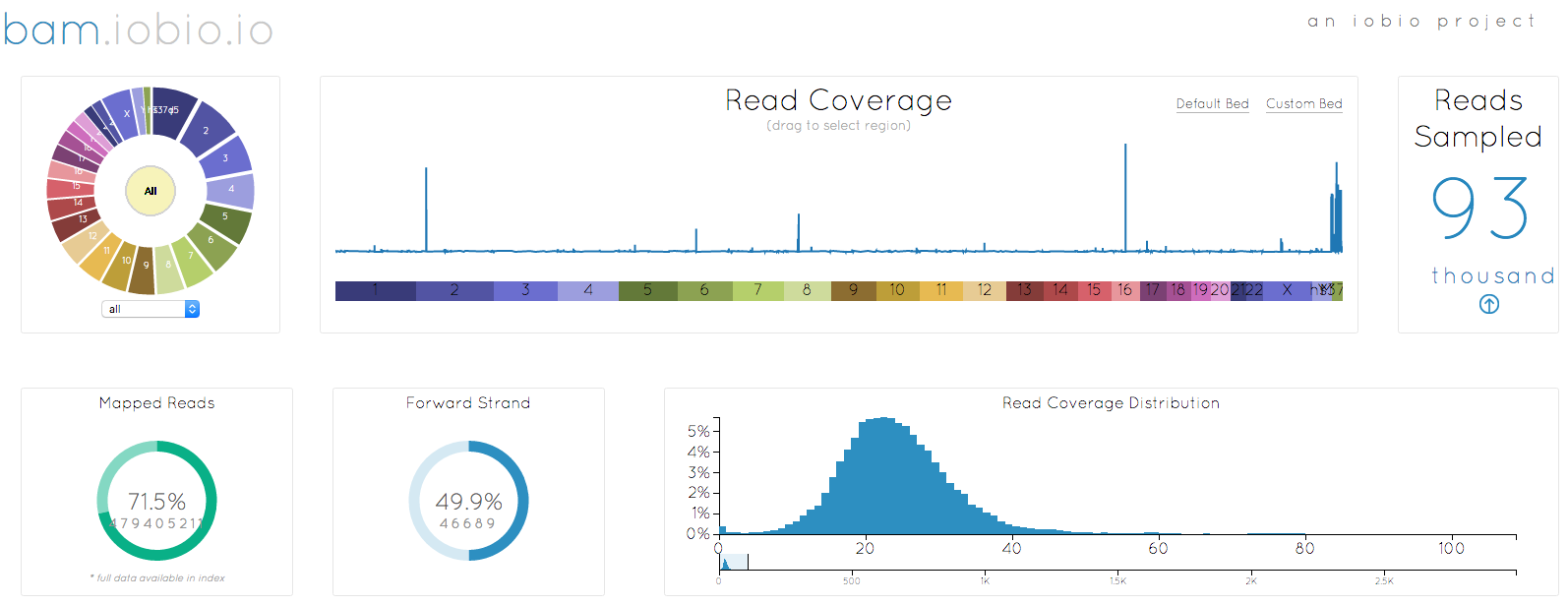
I started out by selecting the bam file to look at (remember that when selecting a local file, you need to select both the bam and the bai index file together), in this case, my own genome. After a few seconds, enough data has been sampled to generate the image at the top of the page. There are a couple of noticable changes to the appearance. First, the chromosome wheel has been added in the top left corner, mirroring the reference sequence selection already implemented in the vcf.iobio. This wheel makes chromosome selection, quick and easy, but also just looks great!!

Another big change is the way that bam.iobio now operates. Sequence reads are sampled from the whole genome and the index file is used to determine the fraction of reads that do not map anywhere to the reference sequence. Note that index files created with older versions of samtools do not have the necessary information to provide this information. These reads are actually really important to help us understand the bam file we are looking at. In a perfect sequencing experiment, all of the reads originated in the genome being studied and they should all map back to the reference genome, leading to a mapping rate of 100%. We know that this never happens though, since the sampled genome does not look exactly like the reference genome. Reads coming from larger insertions, for example, will not map to reference and so will enter the BAM file as unmapped reads, bring down the mapping rate. Even accounting for these differences, we expect the mapping rate to be high, over 90%, so it is a little concerning that only 71.5% of the reads associated with my genome map to the human reference. Maybe I am a more evolved being, with lots of unique DNA, but I suspect that it is actually a result of how the DNA was sampled or problems in the sequencing experiment. Having identified a problem, I can now move to another iobio app, taxonomer.iobio, to take a look at what DNA is present in my BAM file.
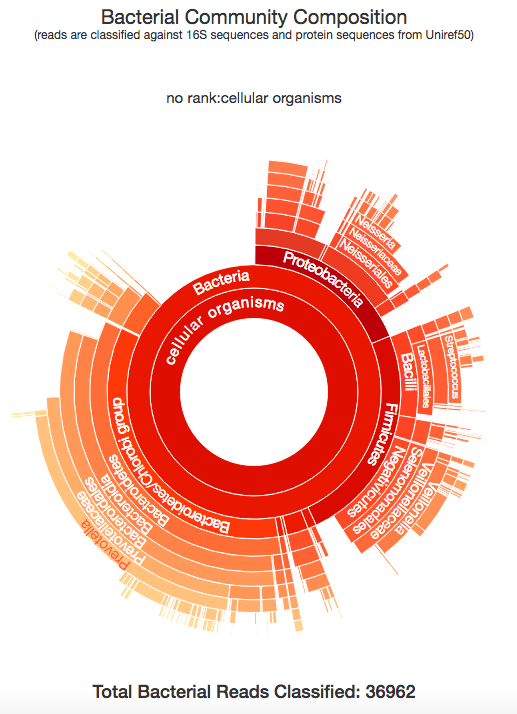
Taxonomer.iobio, and the related commercial version use the Taxonomer software developed in the Yandell lab at the University of Utah, to classify the metagenomic make up of a DNA sample. The image above shows the bacterial composition of the raw DNA reads generated from my sample, generated in a few seconds. This is really interesting and let's me see all of the different organisms that were present in the DNA that is associated with me. All of these bacteria are known to exist in the human microbiome, specifically, the kind of bacteria that are found in a human mouth and mucus. So, what is the conclusion to all of this? By using bam.iobio to look at the quality of my DNA sequencing, I was quickly able to spot a possible contamination problem. Following up on this with taxonomer.iobio, I was able to determine that the DNA was contaminated with DNA from bacteria often found in a human mouth. In a couple of minutes, I identified that my DNA was harvested from a spit sample, rather than from blood. Since I know that I used a spit kit, I know this is the correct conclusion! But, if the source of the DNA you're working with is unknown, these iobio tools give you a very quick way of finding out.


