
A previous post in the gene.iobio version 2 blog series focused on analyzing multiple genes simultaneously. This functionality has really increased the power of gene.iobio, making it a lot easier to analyze a set of genes and identify candidate disease variants. The problem is, though, that we still need to generate a list of genes to analyze. As described in the previous post, we can start with the list of 56 genes identified by the ACMG, but what would be ideal, would be start with a phenotype and generate a gene list from there. Fortunately, that is exactly what we can do!
In this post, we will use the modified Platinum trio dataset and assume this data originates from a family trio where the child is suffering from lactic acidosis. If we know a lot about this phenotype, we may know of some likely genes where we can start our analysis, but what if we do not? Or what if these genes turn up no candidate variants? Using Phenolyzer, a tool developed in the lab of Kai Wang at USC, we can use the phenotype to generate a gene list. For more information on this tool, or other software developed in this lab, go here.
To use Phenolyzer, click on the 'Genes' button to the left of the gene.iobio screen and then enter your phenotype terms where indicated.
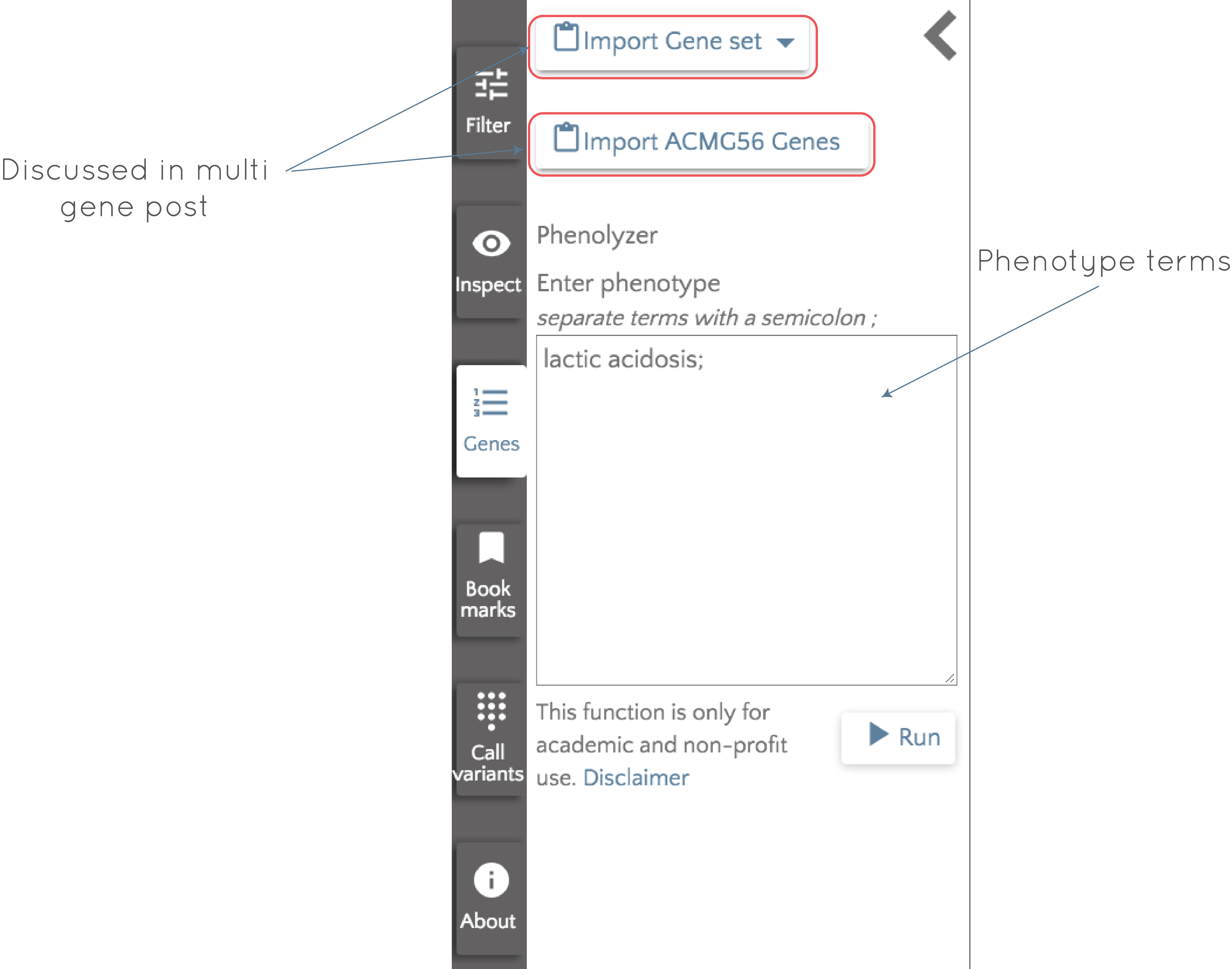
In this case, we enter lactic acidosis, click 'Run' and wait for Phenolyzer to do its magic! Very briefly, Phenolyzer works by trying to match the entered phenotype terms to disease and phenotype terms in its own database to produce a list of seed genes. These genes are then checked for interactions with all other genes in the genome, accounting for four types of interaction; protein-protein interactions, genes in the same Biosystem or the same gene family and in transcription interactions. Based on all of this, a machine learning model is used to generate a score for each gene, and the output is a list of genes ordered by their scores. The output gene list for lactic acidosis appears in the left panel and the top 10 (this number is user configurable) genes appear as gene badges, ready to be interrogated.
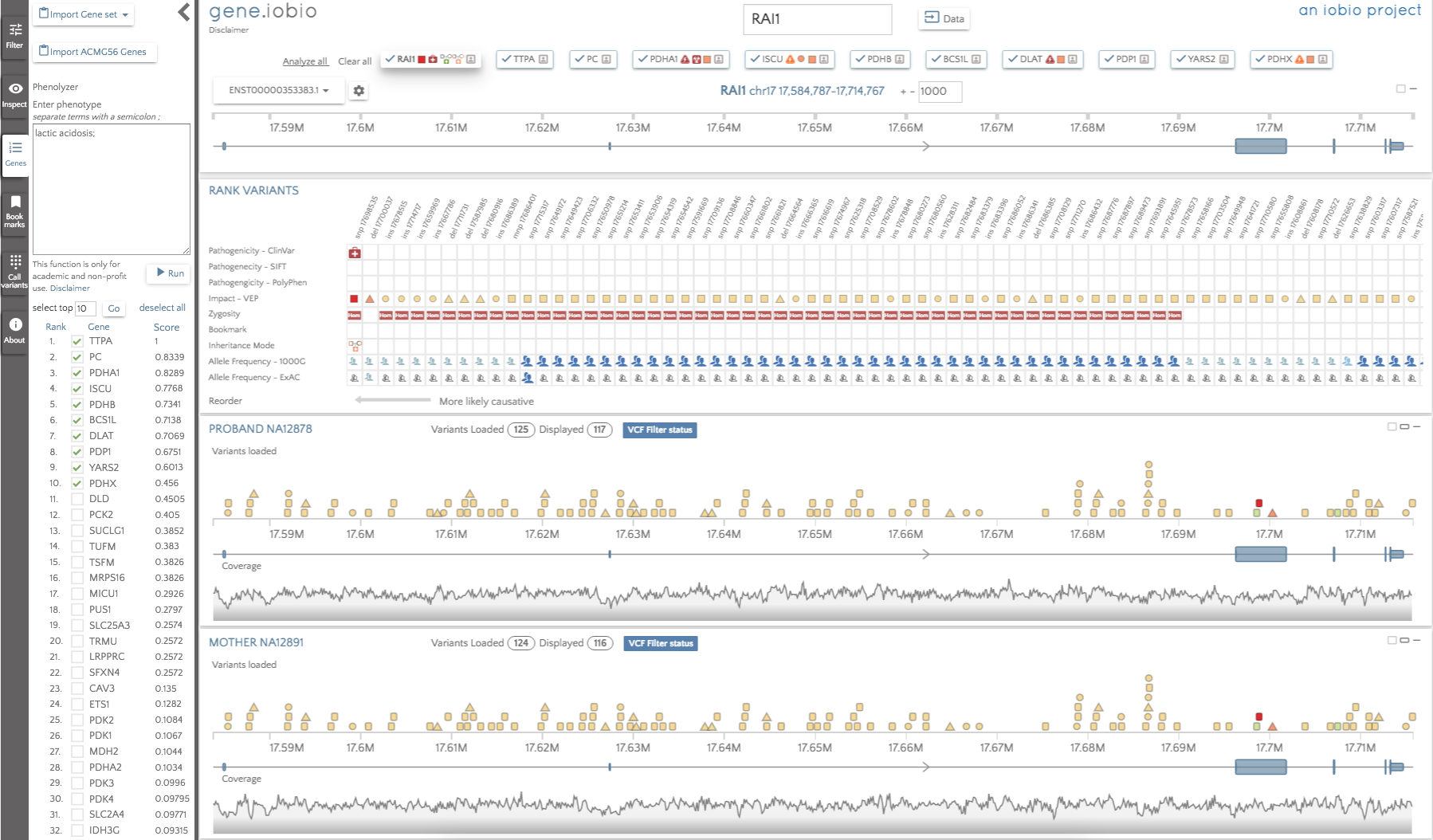
By default, the genes are not automatically analyzed, but we can select 'Analyze all' to begin analysis. When complete, the gene badges describe any interesting variants within them using the glyphs described in the multi-gene analysis post.

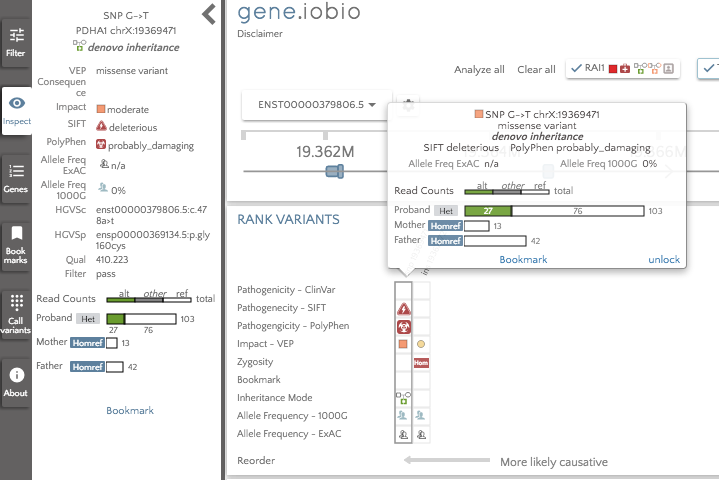
The gene badges are in the prioritized order, so the gene to the furthest left (TTPA) is considered the most important by Phenolyzer. The RAI1 gene still appears in the gene badges, since this was present prior to running Phenolyzer. We can see that there are no glyphs associated with the TTPA gene, so there are no high impact variants to look at there. The highest ranked gene that does have interesting variants to look at is PDHA1, so that is the first gene we should look at. In this gene, we see only two variants; an insertion in a promoter and a de novo missense variant predicted to be deleterious by SIFT and probably_damaging by PolyPhen. The variant is not seen in either 1000 Genomes or in ExAC, so this variant may well be a good candidate for the obseved phenotype.
We hope that you find this functionality useful and we would like to thank Kai Wang and his lab for allowing Phenolyzer to be integrated into gene.iobio.


