
When we released gene.iobio a couple weeks ago, we had a few requests to add Ensembl’s Variant Effect Predictor (VEP) tool.
It will in a new release soon! @jxchong @brent_p
— Gabor Marth (@MarthGabor) August 5, 2015And as any good PI does, Gabor was quick to sign us up to do it! After taking his Twitter account away, we made good on the promise and officially added VEP support, which gives us (along with SnpEff) two powerful variant effect predictor tools.
VEP Consequence
VEP determines the variant consequence on protein coding, a classification that is analogous to SnpEff’s Effect. In fact, these classifications are highly concordant when evaluating coding variants, but keep in mind that these classifications do diverge in the categories of splicing, frameshift, and stop gain. Andrew Jesaitis has written a terrific blog post that explains these differences in his in depth comparison of SnpEff, VEP and Annovar.
We only color variants with either SnpEff or VEP classifications at any given time. You can easily alternate between SnpEff and VEP classifications in the filter panel using the Impact dropdown.
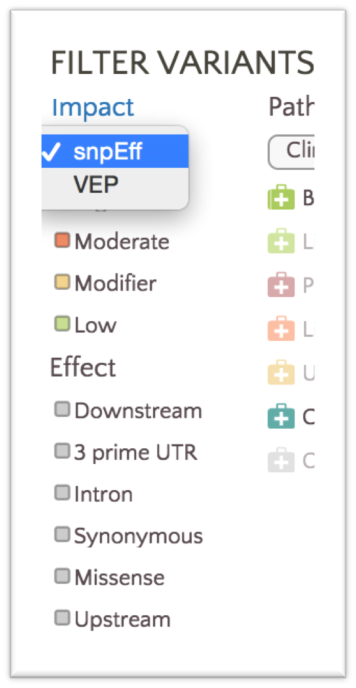
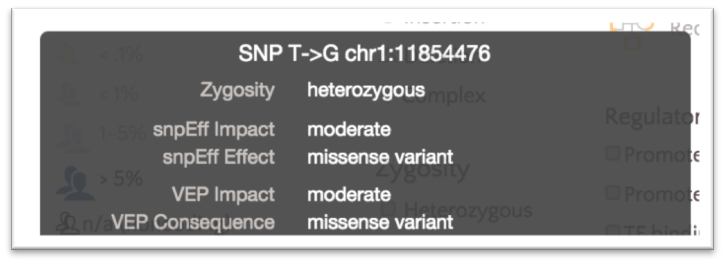
However, both classifications are visible on the variant tooltip displays.
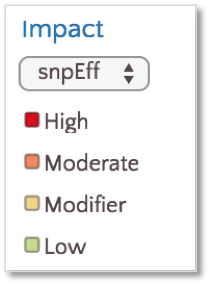
By default, variants are colored based on SnpEff Impact, a high level prioritization scheme on SnpEff Effect, summarizing the categories into four possible levels: High, Moderate, Modifier and Low.
To color based on SnpEff Effect, click on the Effect link.

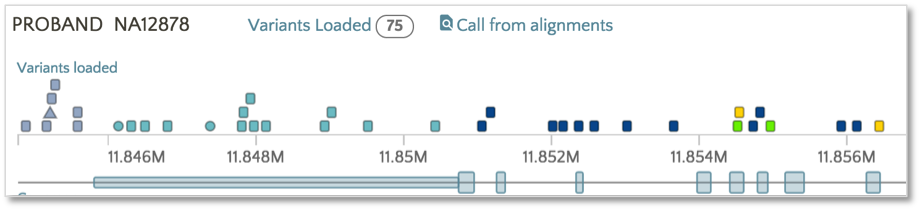
Now the variants have switched from the SnpEff Impact color scheme (e.g. High, Moderate, Modifier, Low) to the SnpEff Effect color scheme (e.g. missense, frameshift, etc.). The same color scheme switching can be performed on VEP impact and consequence.
Predicting Impact on Protein Function
Given that non-synonymous SNPs comprise over half of the mutations known to be involved in human inherited diseases, algorithm-based approaches to predicting disruption to protein structure and function are critical to understanding human disease and phenotype (Wu, Yang 2012). VEP reports classifications from two such algorithms – SIFT and PolyPhen. Both SIFT and PolyPhen analyze sequence homology, whereby substitutions at highly conserved regions are predicted to have higher impact on protein function. In addition, PolyPhen utilizes sequence annotation to inventory features mapped to the location of the substitution. PolyPhen also maps to the 3D protein structure to predict if the amino acid substitution will disrupt the hydrophobic core, alter electrostatic interactions, or interfere with ligand actions.
Gene.iobio provides filters based on SIFT and PolyPhen classifications:


In addition, PolyPhen and SIFT classifications are used to rank the variants.
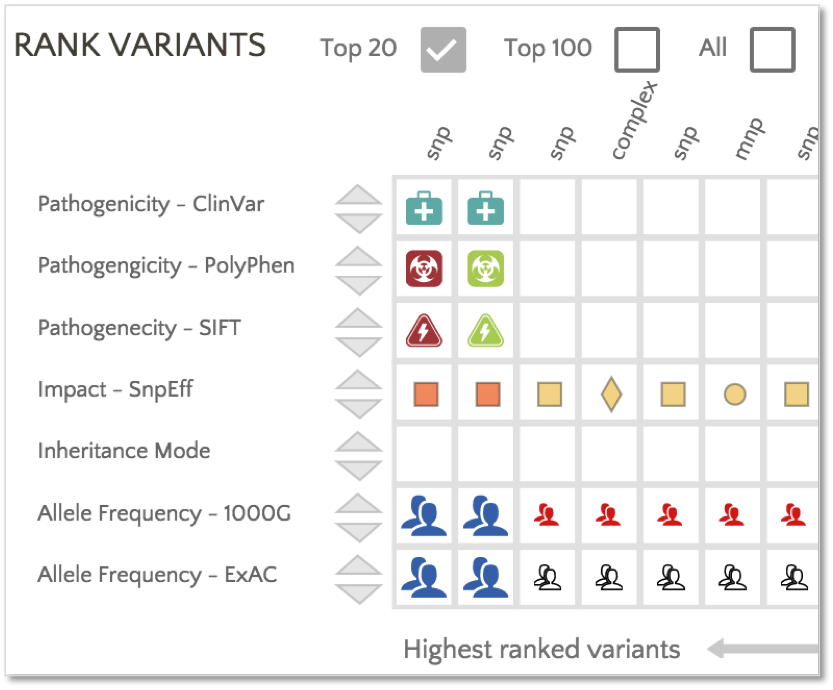
In this example, the first variant in the ranked list has a PolyPhen classification of ‘Probably Damaging’ and a SIFT classification of ‘Deleterious’, providing additional insight into a moderate impact variant catalogued with conflicting interpretations in ClinVar.
When evaluating a variant’s pathogenicity, it is important to consider the variation type and seek other evidence-based sources in addition to the SIFT and PolyPhen predictions. In the paper by Flanagan, et al 2010 which tested the predictive value of SIFT and PolyPhen, it was found that the sensitivity was reasonably high, but that the specificity was quite low, especially for gain-of-function mutations. When considering just SNPs or loss-of-function mutations, these tools performed much better.
Regulatory Annotations
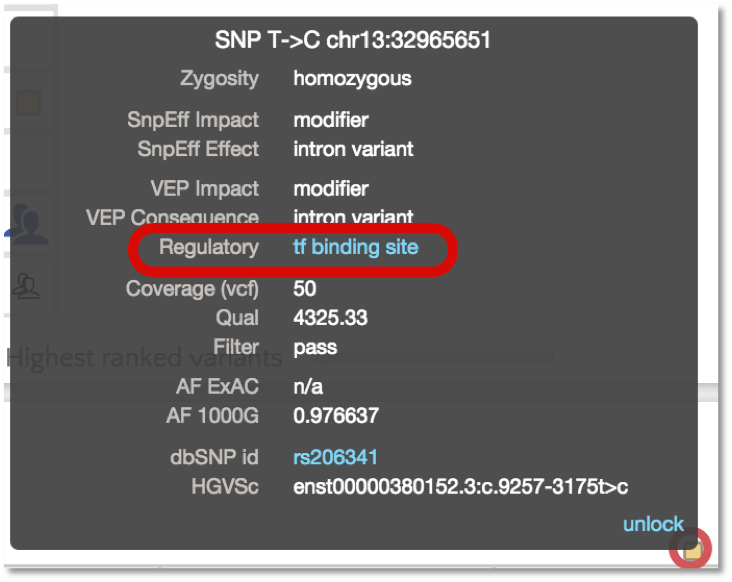
In addition to annotating protein coding variants, VEP also reports on variants that are located in predicted regulatory sites based on the Ensembl Regulatory Build. Gene.iobio now allows variant filtering on these regulatory classifications:

And the variant tooltip provides link outs to Ensembl, showing multiple tracks for different cell types. For example, SNP located in a predicted TF binding site is only active in the Human Embryonic Stem Cell type.
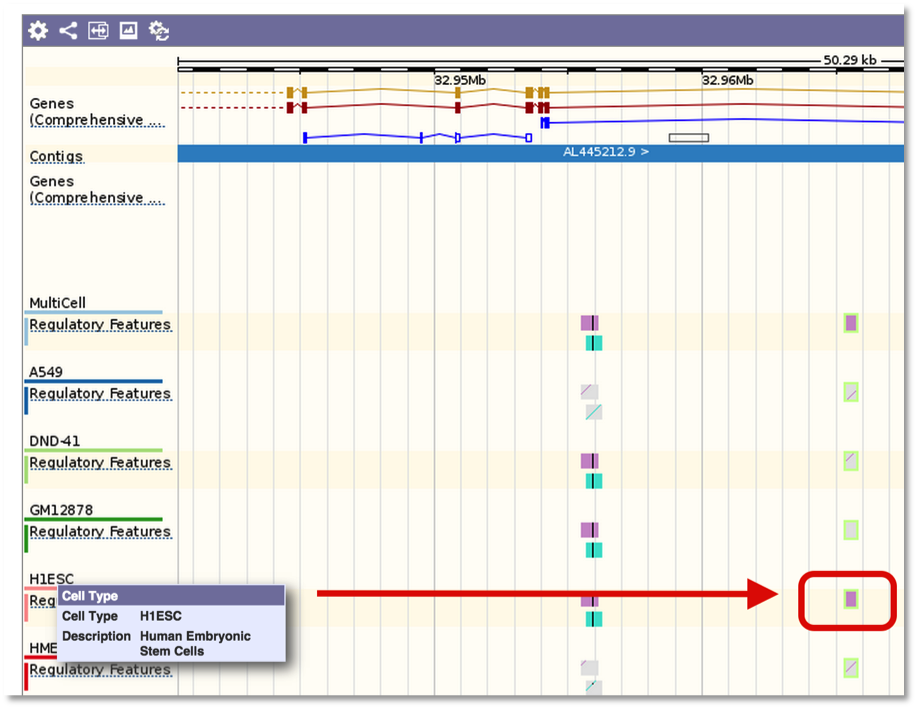
For TF motif features, the link out to Ensembl shows the sequence logo along with a frequency matrix from the JASPER core database (Mathelier, et al 2013).
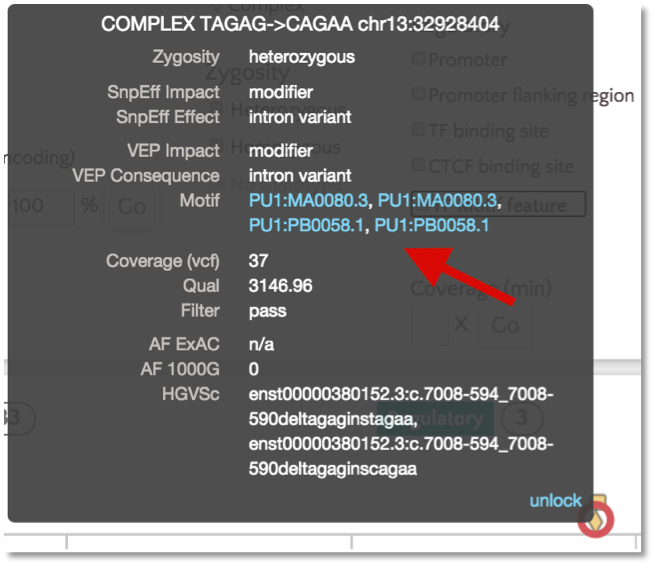
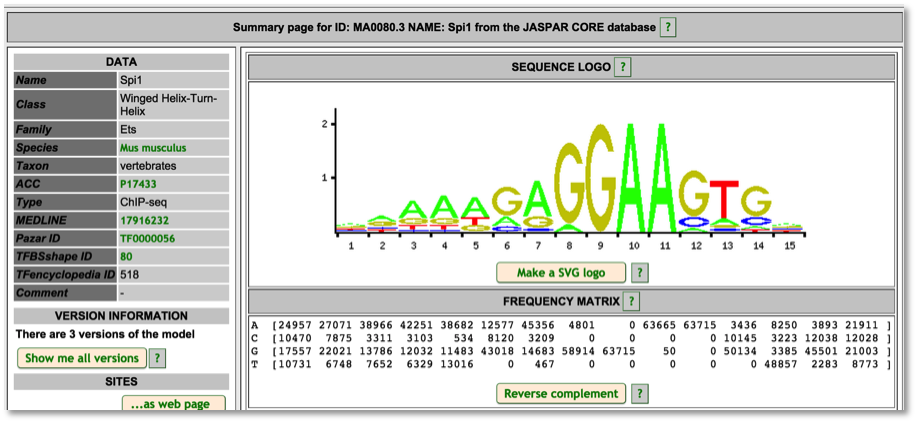
Variant Identifiers and Nomenclature
VEP also provides the dbSNP identifier (i.e. rs number) as well as the HGVS notation for the variant. The HGVS notation is based on the GENCODE transcript set. Additionally, we plan to incorporate the RefSeq transcript set in the near future. In gene.iobio, the tooltip for the variant will show both HGVS notation and the dbSNP id, with the latter linking out to the dbSNP webpage for the particular variant.
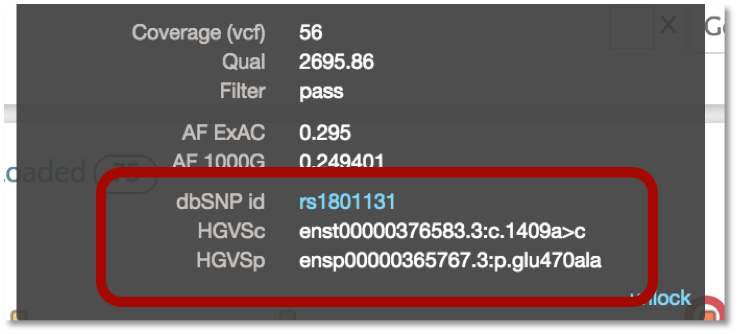
Coming Soon
With VEP integration complete, stay tuned for enhancements to easily navigate through a set of genes, support for multi-sample vcfs, color exons based on sequence coverage, and much more. (VEP is no longer planned!)


