
A new version of gene.iobio is now up and running, featuring several improvements that are another big step forwards!
Outputting variants
The "bookmarks" functionality lets us store variants that we are interested in, so that we can easily jump back to them whenever we want. We heard from many users that they want to be able to output these variants, either to a VCF file, or to an Excel spreadsheet. We now support this functionality. If you select a variant (either by clicking on a variant, or a column in the "Ranked variants" table), a tooltip appears showing a whole host of information about the variant. In the bottom left corner of this tooltip, we can click to "bookmark this variant". This variant is now stored. By clicking on the "Bookmarks" tab at the top of the page, we can look at a summary of all bookmarks.
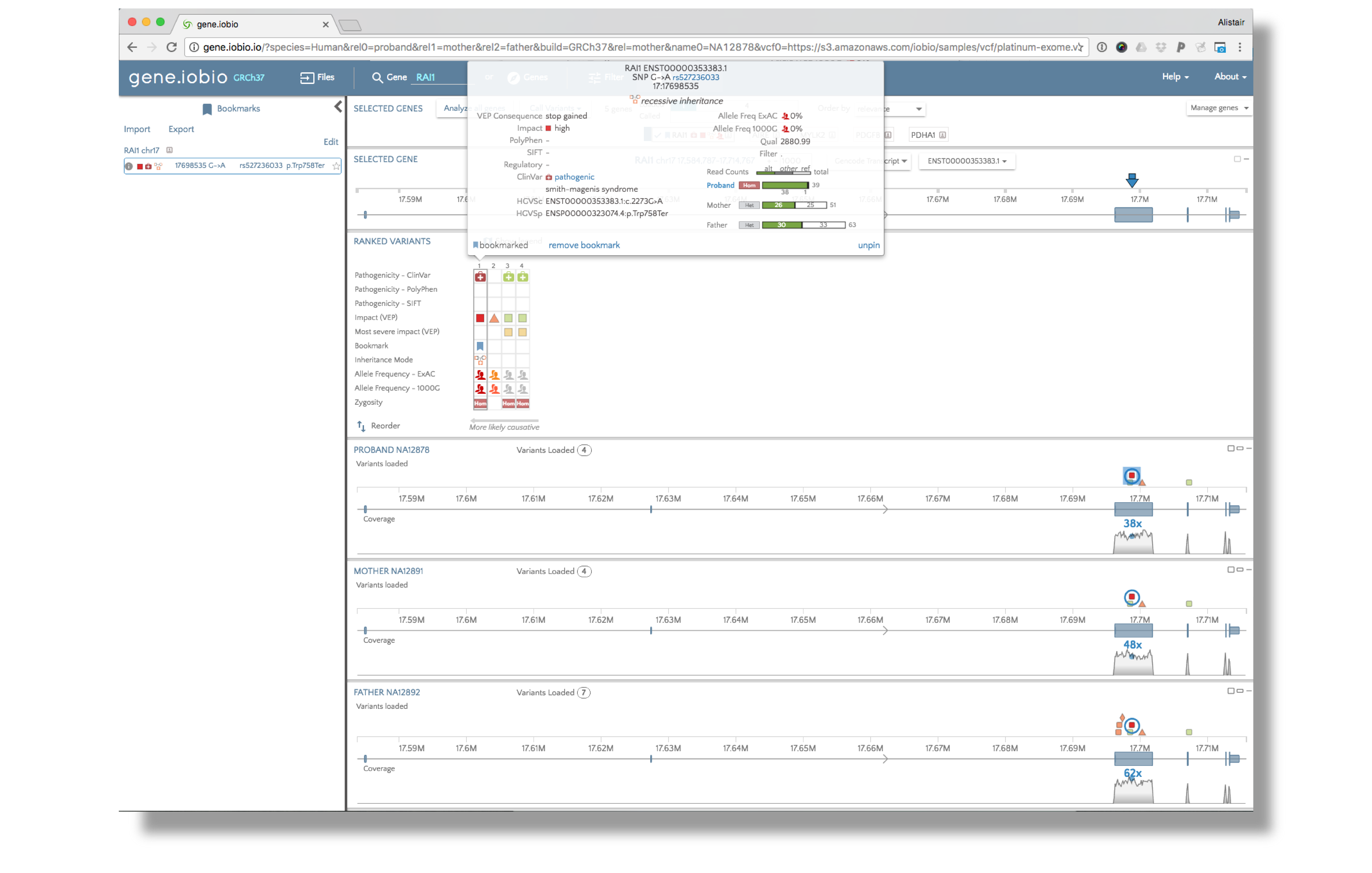
Using the "Export" option, we can choose to export the bookmarked variants as either an Excel, or a VCF file.
Whichever option you choose, the output file contains a list of all the variants, along with all of the annotation information that was available within gene.iobio.
Calling variants
We have removed the "Call variants" tab from the top of gene.iobio and instead, integrated calling into the "Selected genes" panel at the top of the screen. There have also been a number of improvements to the user interface, that we think really improve the user experience.
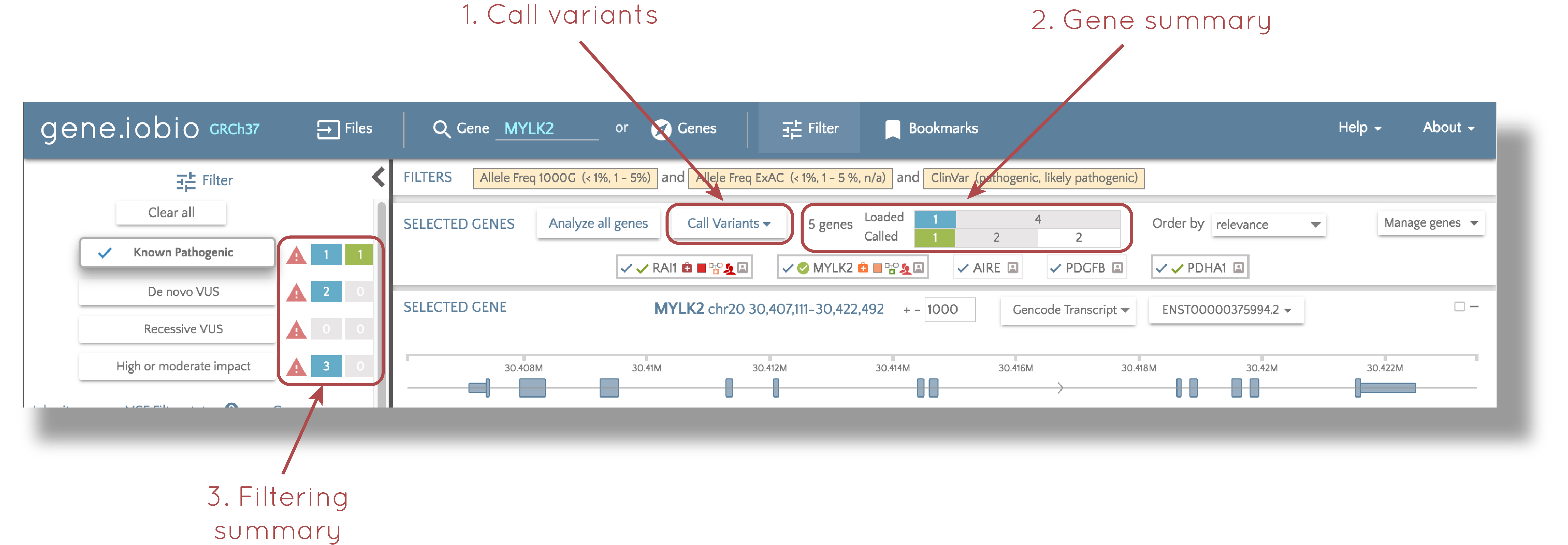
1. Which genes to call variants for?
In the "Call variants" dropdown you can choose to call variants in the gene that you are currently looking at, or you can choose to call variants in all the genes in your list. Once selected, gene will start calling variants.
2. Calling progress
The coloured bars keep track of how many genes have been analyzed, and how many have had new variants called using Freebayes within gene.iobio. In the demo data example above, there are five genes in the list. The top (blue) bar shows how many genes have had the "loaded" variants, i.e. those in the selected VCF file, analyzed, and the lower (green) bar shows how many genes have had variants called. These bars are explained in more detail in the Filtering variants section below.
The gene badges also contain checkmarks to make it clear what analyses have occurred. In the above example, the loaded variants have been analyzed, but no variant calling has happened for the AIRE and PDGFB genes. Both these genes have the check mark ( ) that indicates this. RAI1 and PDHA1 have two check marks (
) that indicates this. RAI1 and PDHA1 have two check marks ( ). The blue check mark indicates that the loaded variants have been analyzed and the green one, that variants have been called. Finally, MYLK2 shows
). The blue check mark indicates that the loaded variants have been analyzed and the green one, that variants have been called. Finally, MYLK2 shows  . Again, the loaded variants have been analyzed, but the called check mark is now in a green circle. This check mark is used to identify when uniquely called variants are present in the gene. The simple green check mark, by contrast, means that variant calling has taken place, but no unique variants were discovered.
. Again, the loaded variants have been analyzed, but the called check mark is now in a green circle. This check mark is used to identify when uniquely called variants are present in the gene. The simple green check mark, by contrast, means that variant calling has taken place, but no unique variants were discovered.
3. Filtering variants
In this example, I have already applied variant filters. The panel at the left shows the results from applying the standard set of filters. Next to the "Known pathogenic" filter, we are presented with three glyphs. The first,  , is provided as a warning to note that not all of the genes have been analyzed. Next we see
, is provided as a warning to note that not all of the genes have been analyzed. Next we see  . This is a count of the number of genes with a loaded variant that passes the filters. Finally, we can see the number of genes with a called variant that passes the filters (
. This is a count of the number of genes with a loaded variant that passes the filters. Finally, we can see the number of genes with a called variant that passes the filters ( ). So in this case, there is one gene with a loaded variant, and one gene with a called variant that counts as a "Known pathogenic" variant. But how do we know which genes these counts are referring to?
). So in this case, there is one gene with a loaded variant, and one gene with a called variant that counts as a "Known pathogenic" variant. But how do we know which genes these counts are referring to?

The gene badges are automatically sorted so that the genes with variants that pass filters appear to the left. So we can see that the furthest left gene badge is for RAI1, followed by MYLK2. Both these genes have other glyphs that indicate properties of the contained variants (see this post for more details), whereas the remaining genes do not. So we know that these are the only two genes we need to look in. Only MYLK2 has the , indicating that variants were called in this gene, so we know that MYLK2 contains the called known pathogenic variant, and RAI1 contains the loaded known pathogenic variant.
Finally, gene provides a visual summary of the analyses that have occured and the filtering results.
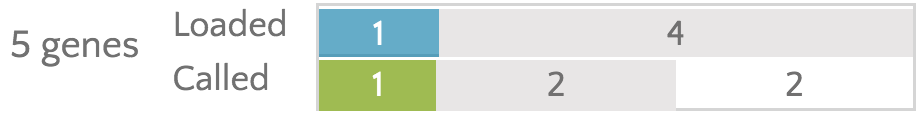
The top bar refers to the loaded variant (remember, these are the variants pulled from the selected vcf file). The bar is split into two. The first segment (in blue) refers to the number of genes that contain a variant passing currently selected filters. In this case, that is one gene. Then we see the remainder of the bar is gray, indicating that the other four genes have been analyzed, but do not contain any variants that pass the filters. The bottom bar works in the same way, but for the called variants. We again see that one gene contains a variant passing the filters. We then see that two genes have had variants called, but no passing variants are present in them (the gray segment). Finally, we see a white segment, indicating that two genes have not yet had variants called. We can confirm this by noting that the AIRE and PDGFB gene badges do not contain the green checkmarks indicating calling has happened.
We hope that these improvements will make it even easier to understand your data and, as always, we encourage you to give feedback on everything iobio related!


