We are really happy to announce the latest release of gene.iobio; version 2.2.0. We have made a major upgrade to the user interface, as well as implementing some new features that really boost the power of analysis with gene.iobio. In this post, I am going to highlight the major changes and recommend that you take a look at the linked tutorials, which demonstrate the power of these new features. The improved filtering functionality significantly enhances gene.iobio, so I strongly urge you read that section! We have also put out a webcast to highlight some of these changes.
New user interface
We have made a number of changes to the basic layout of gene.iobio which we think make it more usable, but also just looks prettier! Here are a couple of the major changes:
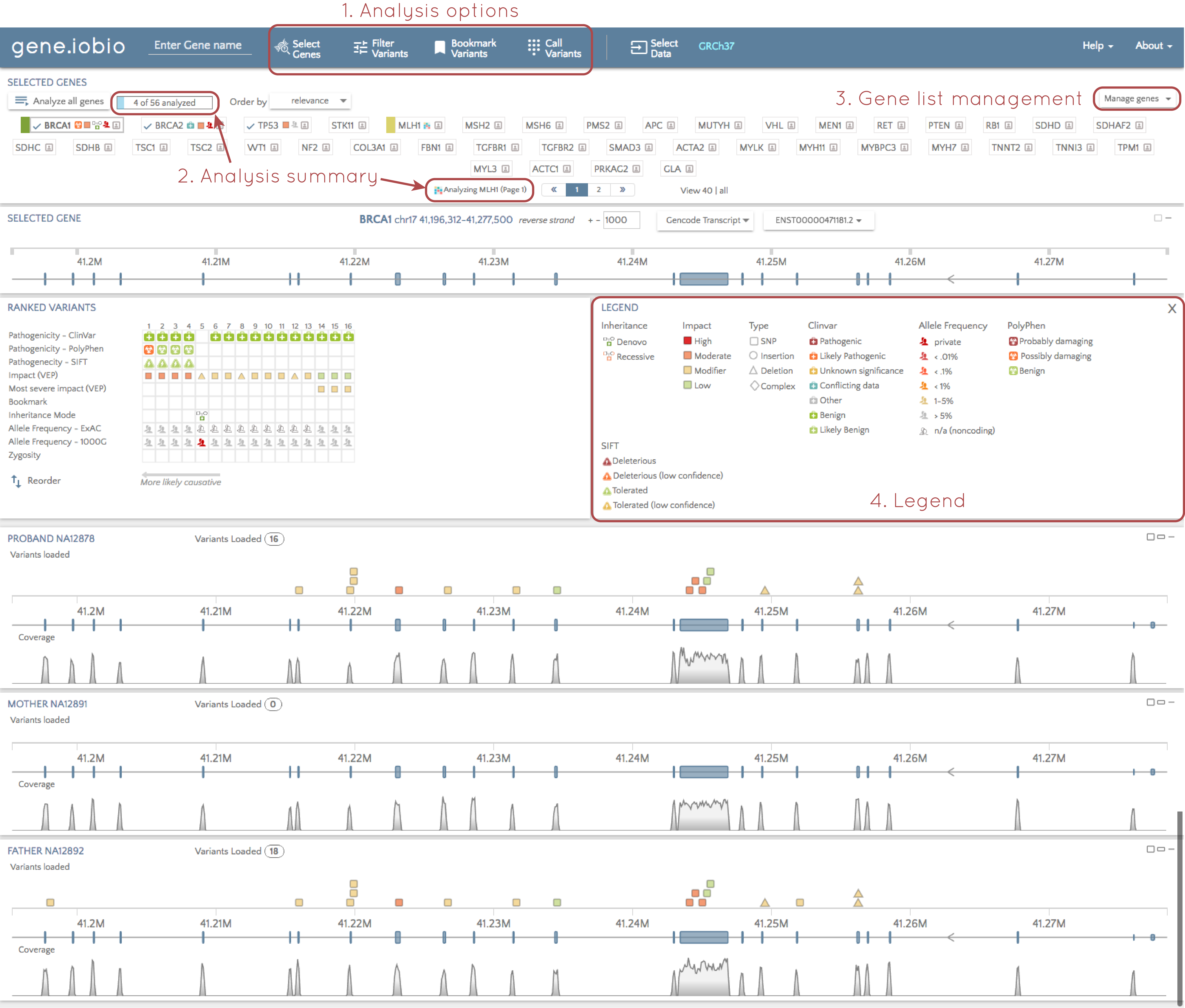
1. Analysis options
We have moved the list of analysis options from the left hand side to the top of the screen. This makes the multitude of analysis options available in gene.iobio more prominent and accessible.
2. Analysis summary
In our own analyses using gene.iobio, we sometimes lost track of how many genes we we were looking and whether or not they were all analyzed. To solve this, we have added a summary of analyzed genes. This provides an up-to-date count of genes we have in our list, and how many of them have been analyzed. If we use the "Analyze all genes" function, gene.iobio will march through all the genes in the list, and update this summary as analysis completes on each gene. In addition, when analysis is underway, we can see which gene is currently being analyzed. This is especially helpful when there are multiple pages of genes, and the gene currently being analyzed is on the next page.
3. Gene list management
We can still import gene lists, or select the ACMG secondary findings genes from the "Select genes" tab at the top of the page, but we now have quick access to modify the gene list on the main page.
4. Legend
We show a lot of information about each variant using lots of different glyphs. If it isn't clear what these glyphs mean, you can now select "Show legend" above the "Ranked variants" table, and a legend describing all these glyphs is shown.
Improved filtering
The modifications to filtering have transformed gene.iobio, making it even easier and more effective in identifying variants of interest. I will use the demo dataset available in gene.iobio to show how powerful the filtering functionality, as well as how easy it is to use. To follow along, open gene.iobio and click "Analyze all genes". This will analyze the variants in each of the genes in turn. When complete, we are ready to start applying filters to identify interesting genes. Click the "Filter Variants" tab at the top of the page to access the filtering functionality.
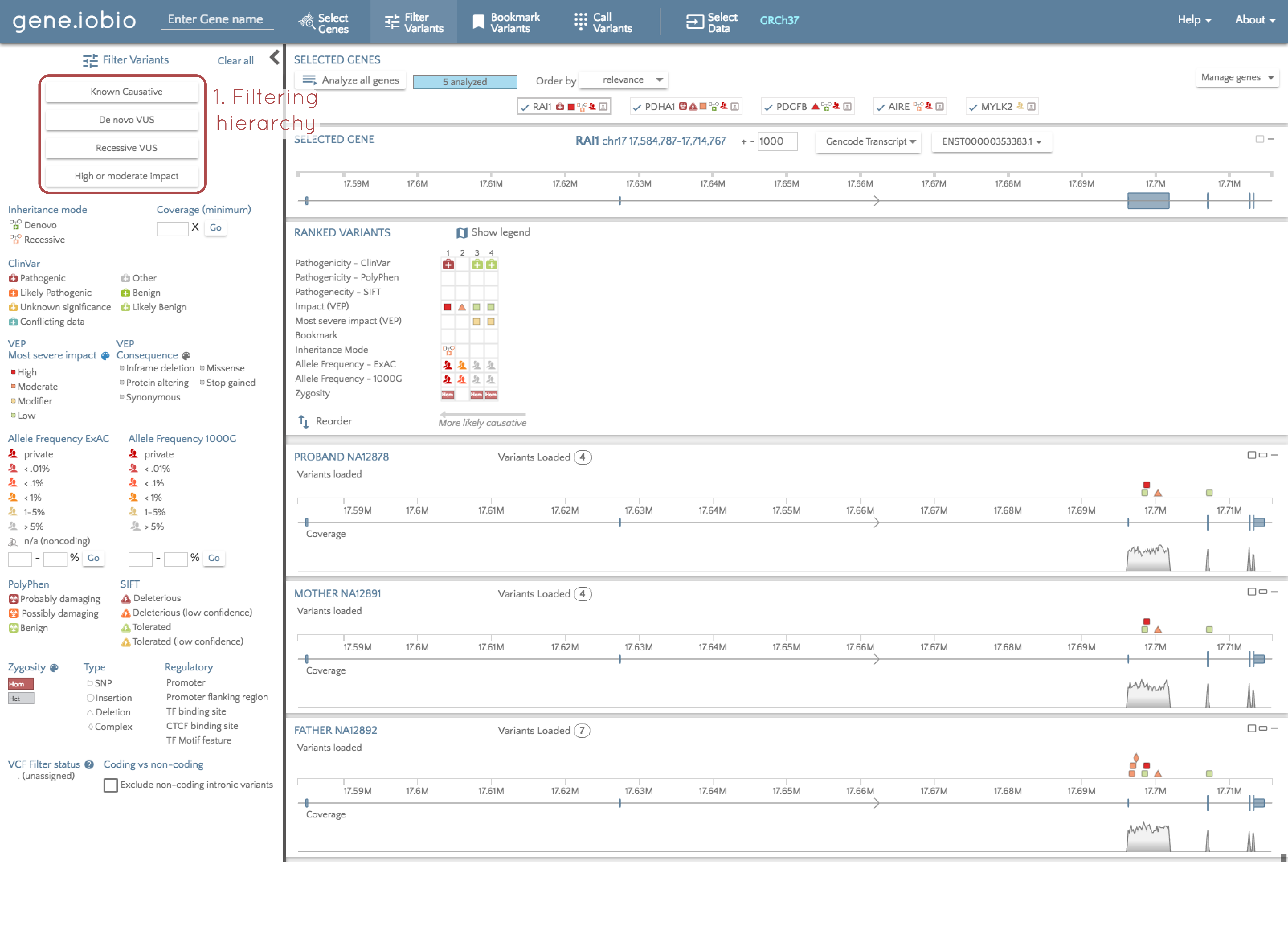
We recommend following a hierarchy based method of variant discovery. By this, I mean that we want to start our analysis by identifying the variants most likely to be responsible for an observed phenotype, and then progressively open up our filters to allow in more and more variants. We can do this using the hierarchy of predefined filters shown in the image above. Starting at the top of this hierarchy, select "Known causative". A few things just happened, so let's look at each of them in turn.
1. Filtering parameters
A set of stringent pre-selected filtering parameters was set to identify rare variants that are known to be pathogenic. We can see what filters were applied in two places (see the image below). In the filter panel at the left, the applied filters are identified with a blue border, and at the top of the screen, the filters are explicitly listed. So, in order for a variant to pass the applied filters, it must be rare (defined in this case as having an allele frequency below 5% in both the 1000 Genomes and ExAC databases), and must be present in the ClinVar database, and annotated as either "pathogenic", or "likely pathogenic".
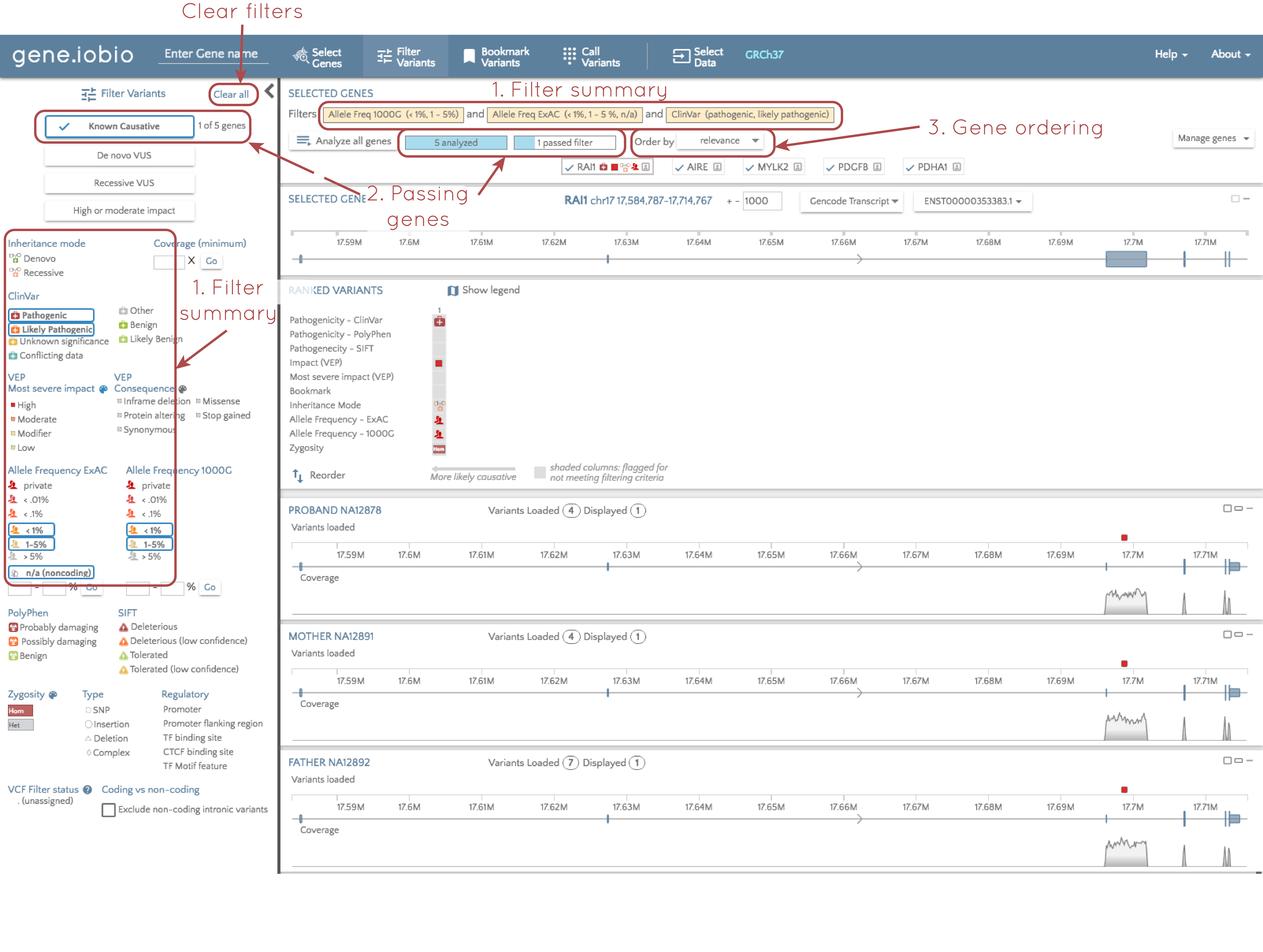
2. Number of genes containing a "passing" variant
When you select a filter, e.g. "Known causative", only genes that contain a variant passing these filters are relevant. To the right of the "Known causative" button, we are told that only one of the five analyzed genes contains a variant that passes these filters. We can also see this at the top of the screen; we are told that all five genes have been analyzed, and the only one gene passes the filters.
If you look at the gene.iobio screenshot under the "Improved filtering" title, each gene in the gene list has a badge at the top of the page, and the gene name is accompanied by a series of glyphs describing contained variants. If you look at the image just above, however, only the RAI1 gene has glyphs. Glyphs are only retained for passing variants, and since RAI1 is the only gene with a variant that passes the filters, this is the only gene that retains glyphs in the gene badge. By comparing the two images again, you can also see that with no filters applied, there are four variants in the RAI1 gene, but after filtering, we are left with a single variant in a single gene that is known to be causative for a disorder.
3. Gene sorting
When you impose filters, we have already seen that the gene list is updated to only show glyphs for genes that contain genes that pass these filters. In addition to the visualization, gene.iobio also reorders the gene badges so that the genes contained passing variants appear first; the genes are ordered according to the functional relevance of the contained genes.
De novo VUS
If no "Known causative" variants are found, or they are ruled out, we can move to different sets of parameters. For example, we can move down the list to find de novo variants of unknown significance (VUS) by selecting this button. As soon as this is clicked, we see the applied filters change - note that ClinVar's pathogenic and likely pathogenic values have a strikethrough. We are not showing any variants that fall into this category as we don't want to duplicate the variants already seen in the "Known causative" category. We see that there are two genes, PDHA1 and PDGFB, that contain de novo VUS variants, and that the list of genes is reordered so that these genes appear at the start of the list. The variant in PDHA appears first since the variant in this gene has more serious Sift and Polyphen scores. Clicking on the gene, moves you to the gene so you can manually look at the contained variants.
Manual filtering
If you work through the preset filter categories, and have not identified a candidate causative variant, we can apply our own filters. First, the selected filter set can be removed by selecting "Clear all" at the top of the filter panel at the left of the screen. Then you can apply filters of your choosing and the gene list and count of passing genes will dynamically reorder


