
Continuing the blog series accompanying the gene.iobio version 2 release, we will discuss how to choose between the GENCODE and RefSeq transcript sets and then different gene transcripts within each set. There are a lot of interesting discussions on the effects of using one or the other gene set. For example, this paper has a more detailed discussion on the two sets, and this paper discusses the differences between functional annotations achieved using different software and transcripts.
How do I select a set?
Before looking into some details about the two gene sets, let's first see how to choose which gene set to use.
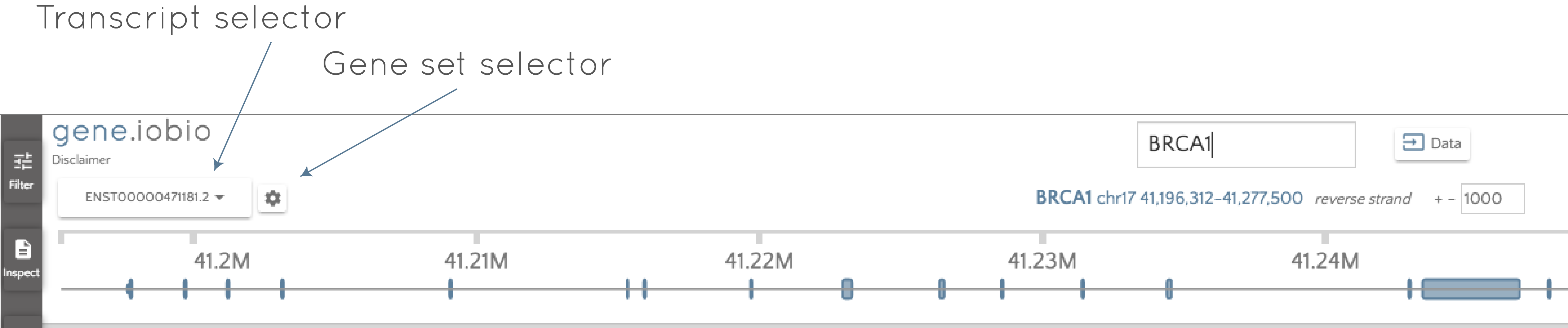
Click the gene set selector wheel to select between the GENCODE and the RefSeq sets and then use the transcript selector to select the desired transcript.
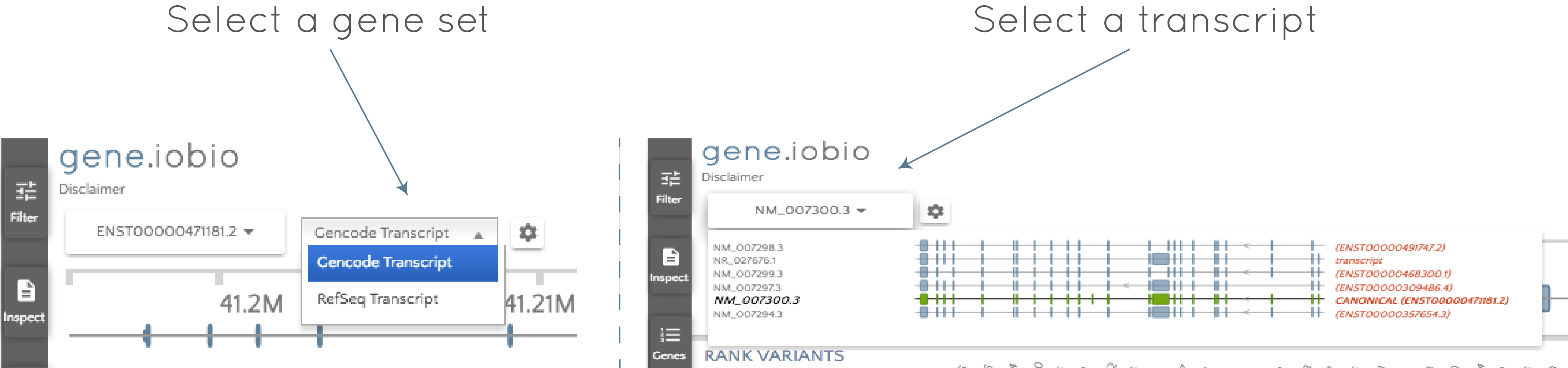
It's that easy! When you change to a different transcript, gene.iobio will reannotate all of the gene variants based on the new transcript. Depending on how different the transcripts are, this can lead to big changes. For example, the PDGFB usecase shows the example of an intronic variant in one transcript being reannotated as a high-impact frameshift mutation in a different transcript.
Some details about the two gene sets
GENCODE gene set
- The GENCODE consortium was set up to provide reference gene annotations for the ENCODE project,
- Aims to be comprehensive, e.g. include pseudogenes, long non-coding RNAs (lncRNAs), short RNAs as well as protein-coding transcripts,
- Includes extensive manual annotation by the HAVANA group, as well as computational annotation. Approximately 93.4% of the annotations involve manual annotation,
- Is undergoing constant validation by many groups in the consortium,
- Is the default annotation set used by the Ensembl project.
RefSeq gene set
- Widely used gene set produced by the NCBI,
- Has significant manually annotated content, but much less than GENCODE (~45% of transcripts are listed as MODEL),
- Transcripts are named as:
- NM: Manually curated, protein-coding transcripts,
- NR: Non-coding transcrips,
- XM: Predicted protein-coding models.
As an example, the available GENCODE and RefSeq transcripts for the BRCA1 gene are shown below. GENCODE clearly has many more available transcripts for this gene. Also, you can see that both gene sets provide the corresponding ID from the other set to the right of the transcript where applicable. The canonical transcript (that picked by default in gene.iobio) is the same in both gene sets (you can see that the GENCODE transcript gives the ID of the canonical RefSeq transcript and vice versa), although the GENCODE transcript includes UTRs, where RefSeq does not.
So which set should I use?
It may be that the your lab uses a particular set of transcripts and has good reason for the choice, so it is probably a good idea to use what your lab/collaborators use. If you want to see more transcript options (including, e.g. pseudogenes), GENCODE would be the better choice for you. In the absense of additional information, we recommend using the canonical transcript.


