
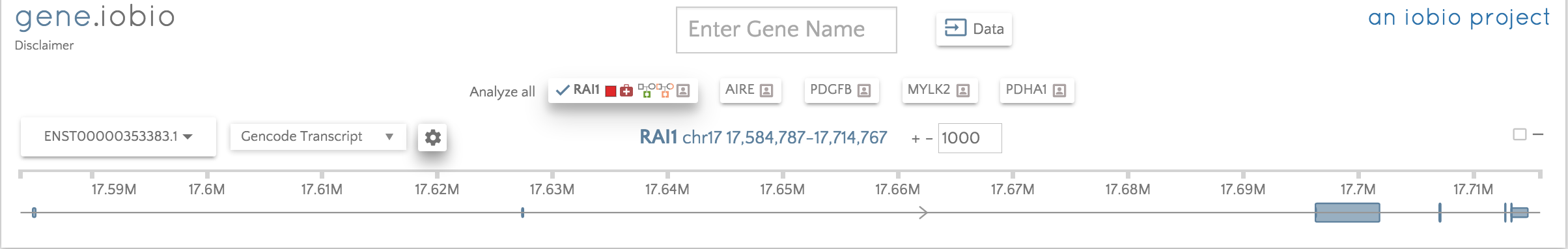
We are thrilled to announce the release of gene.iobio version 2.0! This new release features a major facelift and lots of new functionality that significantly enhances the power and usability of gene.iobio. The single most important enhancement is that we have now moved from being a single gene to a multi-gene assessment tool, with integrated methods of generating and importing gene lists, including manual import and phenotype driven list generation with Phenolyzer. Below, we can see where genes being investigated can be selected and the transcript set can be switched between Gencode and RefSeq.
This post is the first in a series describing the new interface and features and will focus on changes to the interface. The series will then continue to address in more detail, the following topics:
- Displaying variants based on the VCF filter status,
- Using RefSeq and GenCode gene transcripts,
- Analyzing multiple genes with gene.iobio,
- Analyzing the ACMG56 genes,
- Generating gene lists from phenotype terms using the Phenolyzer tool from USC,
- Bookmarking and exporting candidate variants,
- Importing candidate variants from e.g. GEMINI and interrogating with gene.iobio,
- Performing analysis with affected/unaffected siblings.
We have included a series of menu buttons down the left hand side of the screen where all the analysis functionality is now accessible. When a button is selected, a panel is opened containing all the actions or information associated with the selected function. We will not discuss all of the buttons in this post (they will all be covered in the remainder of this series), but will demonstrate one of the panels. The image below shows the tutorial usecase for the RAI1 gene.
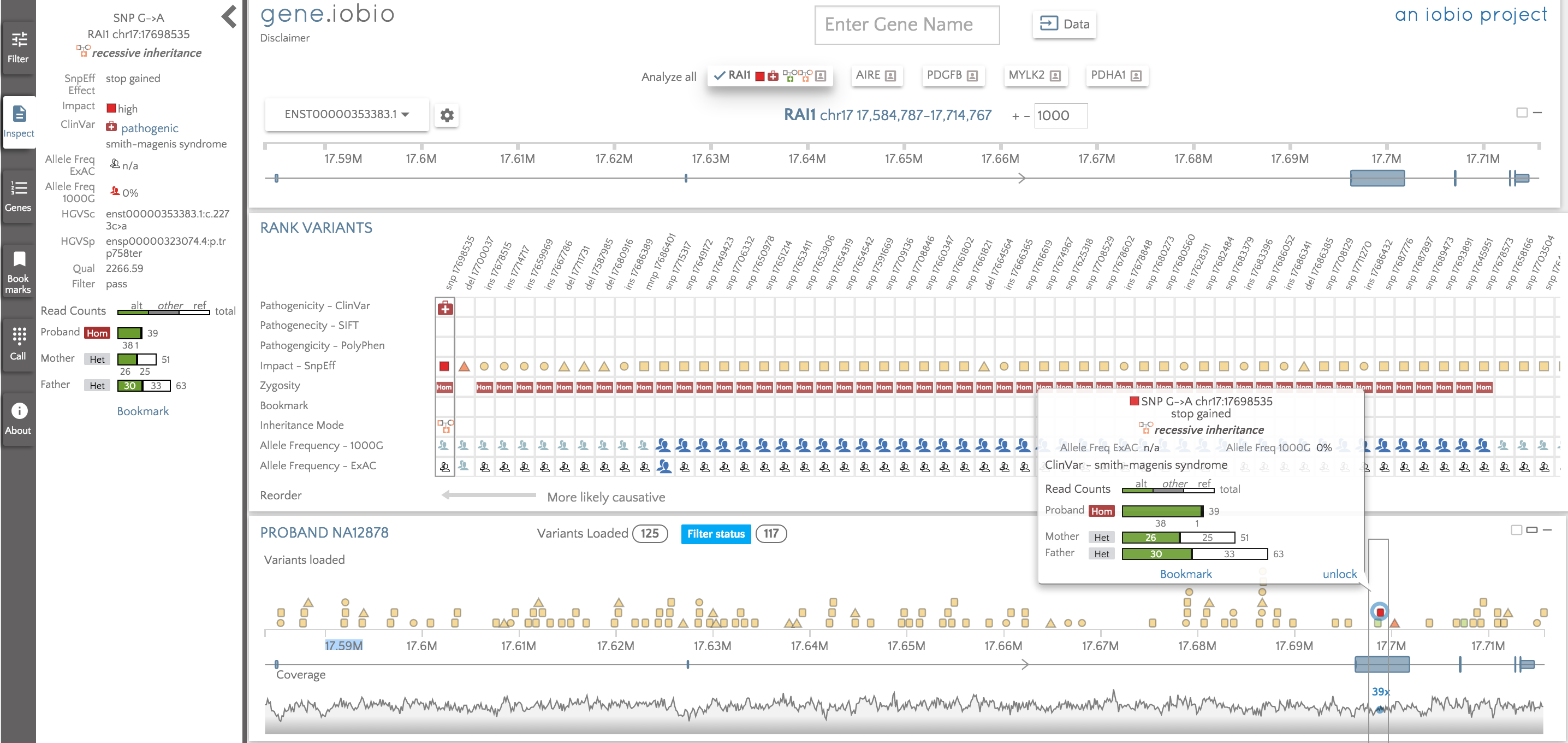
If we mouse over a variant in the proband card, a tooltip appears with limited details about the variant. We have included in this tooltip the allele counts for the reference, alternate and other alleles (in the event of a sample showing more than two alleles due to e.g. error, mosaicism etc.) for all samples. The called zygosity for each of the samples is included next to the counts, so it is really easy to compare the calls across the family. If we want to know more details, click the variant and the left panel immediately jumps to 'Inspect' and is populated with additional details about the variant, including, where applicable, links out to ClinVar, NCBIs dbSNP database etc. We will be continuing to add more information to this panel as time goes by and will strive to address any of your suggestions or comments.
We recommend trying out all the new features for yourself, including the powerful new Phenolyzer tool, accessible by clicking the 'Genes' button and entering a phenotype, for example, 'lactic acidosis'. All the new features will be discussed in this blog series that we will be releasing regularly over the coming weeks. Remember, you can still access all resources, including gene.iobio tours, tutorial use cases, screencasts, blogs etc. by clicking the 'About' button.


